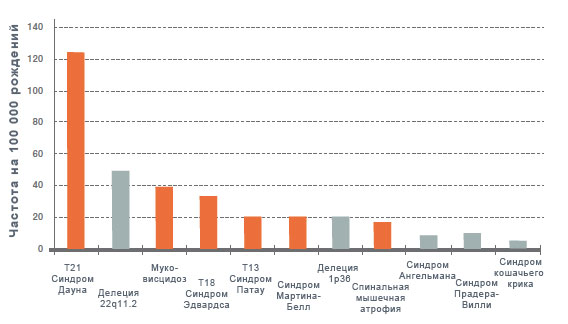
Трисомия 21, трисомия 18 и трисомия 13 — три наиболее распространенных анеуплоидии, которые в общей сложности возникают примерно в 1 случае на 450 живых рожденных детей. Из них наиболее распространена трисомия 21. Клинически важные микроделеции встречаются чаще, чем считалось ранее, и могут возникать при беременности с отсутствием аномалий при УЗИ. Совокупная частота пяти синдромов микроделеций при рождении, охватываемых тестом Panorama, составляет приблизительно 1:1000. Наиболее распространенное значимое состояние микроделеции, синдром делеции 22q11.2, встречается чаще, чем муковисцидоз.
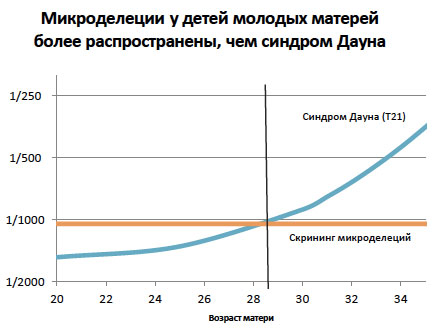
Кроме того, в отличие от анеуплоидий по всей хромосоме (например, трисомии 21), которые встречаются чаще у женщин более старшего возраста, риск микроделеций не зависит от возраста матери. Для беременных женщин моложе 28 лет это означает, что у них более вероятна беременность с одной из этих пяти микроделеций, чем с синдромом Дауна. Наибольшую озабоченность вызывает то, что эти синдромы микроделеций могут быть тяжелыми и способны приводить к серьезным физическим и/или интеллектуальным поражениям, которые в противном случае останутся недиагностированными до детского возраста.
До недавних пор основной вариант скрининга на анеуплоидию включал в себя измерение биохимических маркеров в сыворотке крови матери на первом и/или втором триместре, иногда в сочетании с ультразвуковым исследованием на первом триместре для измерения прозрачности шейной складки (NT). Хотя этот интегрированный скрининговый подход был разработан для выявления трисомии 21, он способен также обнаруживать трисомию 18 и трисомию 13. Повышенная NT (а также кистозная гигрома и другие индикаторы) также связана с определенными анеуплоидиями половых хромосом (и косвенным образом связана с синдромом делеции 22q11.2).
Исследования с использованием традиционных скрининговых методов сообщали о частоте ложноположительных результатов 4–5% при обнаружении трисомии 21. Диагностическая ценность положительногорезультата (PPV) — т. е., вероятность, что результат высокого риска по тесту укажет истинный положительный результат у пациента — для трисомии 21 с использованием традиционных скрининговых методов составляет <5%. Это значит, что для каждых более двадцати женщин, которые получат положительный результат скрининга, по меньшеймере у девятнадцати он будет ложноположительным.

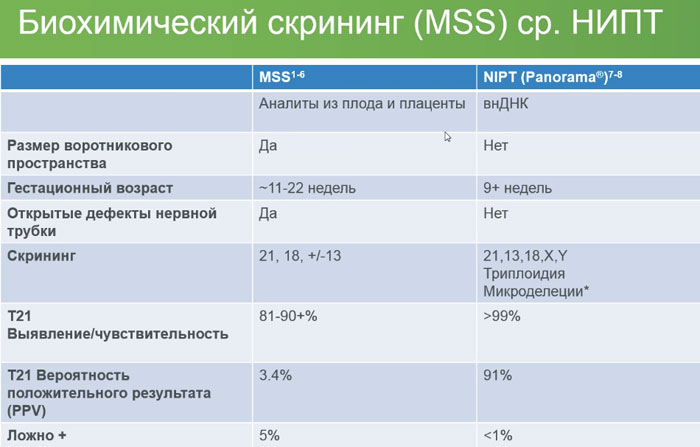
Традиционные скрининговые методы не предназначены для обнаружения микроделеций. В отличие от анеуплоидий, синдромы микроделеции не имеют хорошо определенных маркеров, которые могут быть обнаружены с использованием скрининга сыворотки. При многих из этих синдромов также отсутствуют структурные аномалии, которыемогут быть обнаружены при УЗИ. Пренатальная диагностика синдромов микроделеций требует инвазивной процедуры, т. е. амниоцентеза или биопсии ворсин хориона (CVS), для получения клеток для последующего анализа с использованием флуоресцентной гибридизации in situ (FISH) или хромосомного микроматричного анализа. Процедуры CVS и амниоцентеза, которые включают в себя только кариотипирование (а не FISH или микроматричный анализ), не могут обнаружить большинство микроделеций. Для беременных женщин, проходящих инвазивное тестирование, подходит микроматричный анализ, независимо от возраста матери. Однако, так как с инвазивными процедурами связан риск потери беременности (1/450–1/900), многие женщины не проходят инвазивное тестирование в рутинном порядке, если у них отсутствуют другие указания на высокий риск. Поскольку эффективные неинвазивные методы пренатального скрининга отсутствовали, многим детям с нарушениями вплоть до рождения или даже до взрослого возраста неставился диагноз; в результате, возможности для раннего вмешательства были упущены. Таким образом, имеется очевидная потребность в подходе пренатального скрининга, который является точным и неинвазивным у всех пациентов.
Синдром делеции 22q11.2/Синдром Ди Джорджи
Дети, родившиеся с синдромом делеции 22q11.2, часто страдают пороками сердца, нарушениями иммунитета и легкой или умеренной умственной отсталостью. Они также могут испытывать проблемы с почками, проблемы с питанием и/или страдать эпилепсией.
1. Гипокальциемия. Низкие уровни кальция, или гипокальциемия, часто наблюдаются при синдроме делеции 22q11.2, особенно у новорожденных. Низкие уровни кальция могут приводить к судорогам. Часто бывает так, что причина гипокальциемических судорог остается незамеченной и без лечения, пагубно влияет на умственное развитие ребенка. Необходимо вести контроль за признаками гипокальциемии у новорожденных с делецией 22q11.2, и в случае обнаружения незамедлительно проводить лечение. Гипокальциемия может возникать повторно у пациентов, страдавших ей, при вспышках роста, в пубертате, при болезни или при хирургическом вмешательстве.
2. Иммунодефицит. Почти у 75% пациентов с делецией 22q11.2 имеются проблемы с иммунитетом. В связи с риском иммунодефицита необходимо проводить оценку больных до введения вакцины, содержащей живые вирусы.
3. Патологии нёба. Почти у 75% пациентов с делецией 22q11.2 наблюдаются проблемы с нёбом, структурные, функциональные или структурные и функциональные. Эти проблемымогут приводить к трудностям с питанием и речью. При отсутствии коррекции в раннем возрасте возможно нарушение формирования речи. В большинстве случаев эти состоянияподдаются лечению.
4. Проблемы с питанием. У новорожденных с делецией 22q11.2 часто встречаются проблемы с питанием, не связанные с патологиями нёба или сердца. Первопричина проблем с питанием может быть связана с нарушением моторики глотки и пищевода и часто приводит к рефлюксу и запору. В редких случаях наблюдались мальротация кишечника и болезнь Гиршпрунга. Большинство этих проблем поддается лечению.
5. Врожденные пороки сердца. Почти 75% пациентов с делецией 22q11.2 страдают врожденными пороками сердца; это обнаружение часто является причиной диагноза. Если упациента диагностирована делеция 22q11.2, его следует направить к кардиологу.
6. С делецией 22q11.2 могут быть связаны и другие медицинские проблемы, включая аномалии почек, потерю слуха, осложнения со стороны ЛОР-органов, аутоиммунные заболевания и скелетные аномалии.
Синдром делеции 1p36
Дети, родившиеся с синдромом делеции 1p36, имеют слабый мышечный тонус, пороки сердца и другие врожденные пороки, умственные нарушения и проблемы с поведением. Примерно половина из них страдает эпилепсией.
Синдром Ангельмана
У детей, родившихся с синдромом Ангельмана, часто наблюдается задержка развития моторных навыков (умения сидеть, ползать и ходить), эпилепсияи проблемы с равновесием и ходьбой. Для них также характерна тяжелая умственная отсталость и отсутствие речи.
Синдром кошачьего крика или 5P минус
У детей с синдромом кошачьего крика обычно низкая масса при рождении, небольшой размер головы и сниженный тонус мышц. Также распространены проблемы с питанием и дыханием. Этот синдром связан с умеренной или тяжелой умственной отсталостью.
Синдром Прадера-Вилли
Дети, родившиеся с синдромом Прадера — Вилли, имеют низкий мышечный тонус и проблемы с питанием и набором веса. Для них также характерна умственная отсталость. Как в детском, так и во взрослом возрасте они быстро набирают вес и часто страдают болезнями, связанными с ожирением.
Революцию в скрининге на фетальные анеуплоидии совершили неинвазивные пренатальные тесты, анализирующие вкДНК в плазме крови матери. Поскольку фетальная вкДНК проходит через плацентарный барьер и поступает в кровоток матери, простой отбор крови у матери обеспечивает средства для обнаружения фетальных копий хромосом, не подвергая плод рискам, связанным с инвазивными процедурами. Анализируемая вкДНК представляет собой смесь ДНК матери и плода, процент ДНК плода называется «фетальная фракция». Было показано, что фетальная фракция положительно коррелирует с гестационным возрастом и отрицательно коррелирует с весом матери. Точное определение фетальных копий хромосом с использованием вкДНК, изолированной из плазмы крови матери, требует амплификации вкДНК и последующего биоинформационного анализа. На настоящий момент имеется два основных биоинформационных подхода: «количественные» методы или методы подсчета первого поколения, используемые большинством тестов, основанных на вкДНК, и подход второго поколения, используемый Natera, который включает в себя информацию генотипирования, основанную на однонуклеотидных полиморфизмах (SNP).
Первое поколение: методология подсчета
Количественные методы подсчета сравнивают относительное количество считанных последовательностей от интересующей хромосомы, например хромосомы 21 (трисомия по которой приводит к синдрому Дауна) с эталонной хромосомой или набором хромосом, для которых предполагается эуплоидность. Хотя этот подход эффективен для обнаружения трисомии 21 и трисомии 18, он с меньшим успехом обнаруживает трисомию 13 или анеуплоидии половых хромосом.20-24 Метод подсчета не способен обнаружить триплоидию, которая, по оценкам, возникает приблизительно в 1 случае на 2000 случаев беременности на 12 неделях,25,26 и которая может привести к тяжелым аномалиям плаценты и плода вместе с повышенными рисками спонтанного аборта, пре-эклампсии и гестационной трофобластической неоплазии. Метод учета также не способен различать генотипы матери и плода, что может стать проблемой в случае дупликаций у матери. Этот метод также не дает возможности обнаружения синдрома «исчезающей двойни»; как аномалии матери, так и беременность с синдромом «исчезающей двойни» приводят к ложноположительным результатам для плода.27-30 Так как фетальная фракция вкДНК на самом деле происходит из плаценты, существуют случаи, когда фетальная и плацентарная ДНК различаются вследствие ограниченного плацентарного мозаицизма. Этот тип мозаицизма возникает при 1–2%беременностей31-33 и может являться причиной ложноотрицательных и ложноположительных результатов как для методологии подсчета, так и для методологии, основанной на SNP.
Второе поколение: методология, основанная на SNP
Тест Panorama является единственным доступным подходом NIPT, специально нацеленным на SNP для определения плоидности. Этот подход выделяет ДНК матери из ее лейкоцитов, осуществляет секвенирование и использует эту информацию для «вычитания» генотипа матери из образца плазмы. Это обеспечивает более надежные данные о генотипе плода и более высокую точность даже при фетальных фракциях не более 2,8%. Тест Panorama направлен на 13 392 SNP, охватывающих хромосомы 21, 18, 13, X и Y; дополнительные наборы SNP являются целью для обнаружения микроделеций. Затем используется запатентованный алгоритм для скрининга на фетальную хромосомную анеуплоидию и пол плода. Способность различения вкДНК матери и плода также позволяет Panorama идентифицировать синдром «исчезающей двойни» и дупликации матери и может привести к идентификации ранее не выявленной микроделеции, например синдрома делеции 22q11.2 у матери. Так как для теста Panorama не требуется эталонная хромосома, он уникальным образом способен обнаруживать триплоидию и полностью молярную беременность.
Валидационное исследование на хромосомы 13, 18, 21, X и Y показало значения чувствительности 100% для трисомии 21 и трисомии 13, 96% для трисомии 18 и 90% для моносомии X; по каждому показанию к применению были получены значения специфичности не менее 99,9%.2 Значения диагностической ценности положительного результата (PPV) для результата «высокий риск» были определены после крупного клинического валидационного исследования Panorama.1 PPV в этом исследовании составили 90,9% длятрисомии 21, 93,1% для трисомии 18, 38,1% для трисомии 13 и 50,0% для моносомии X.1 В валидационном исследовании микроделеций34 сообщались значения чувствительности более 97% и значения специфичности более 99% для каждого состояния микроделеций. Клиническая валидация обнаружения микроделеций продолжается в настоящее время.